
Published by Zhuhai Biori Biotechnology Co., Ltd.
Recent discussion around FDA Form 483 response expectations is a reminder that regulated life-science manufacturing depends not only on good science, but also on complete, review-ready documentation. For molecular diagnostics and IVD reagent programs, IVD reagent quality documentation helps connect raw materials, manufacturing records, QC release data and customer technical files into one traceable system.
This article translates the current regulatory documentation conversation into practical supplier-selection questions for diagnostic assay developers, OEM partners and reagent sourcing teams.
Why IVD Reagent Quality Documentation Is Getting More Attention
Fierce Pharma recently reported on industry questions around the FDA’s draft guidance for responding to Form 483 observations after drug CGMP inspections. The guidance is focused on drug CGMP inspection responses, not IVD reagent suppliers specifically, but the broader signal is relevant: regulators and customers expect timely evidence, root-cause thinking, corrective actions and records that can be followed from process to batch release.
For IVD raw materials and molecular diagnostic reagents, documentation quality can influence supplier qualification, lot acceptance, technical transfer, deviation handling and long-term OEM collaboration. A strong reagent may still create supply-chain risk if the supporting records are incomplete, inconsistent or difficult to audit.
What an IVD or OEM Reagent Documentation Package Should Cover
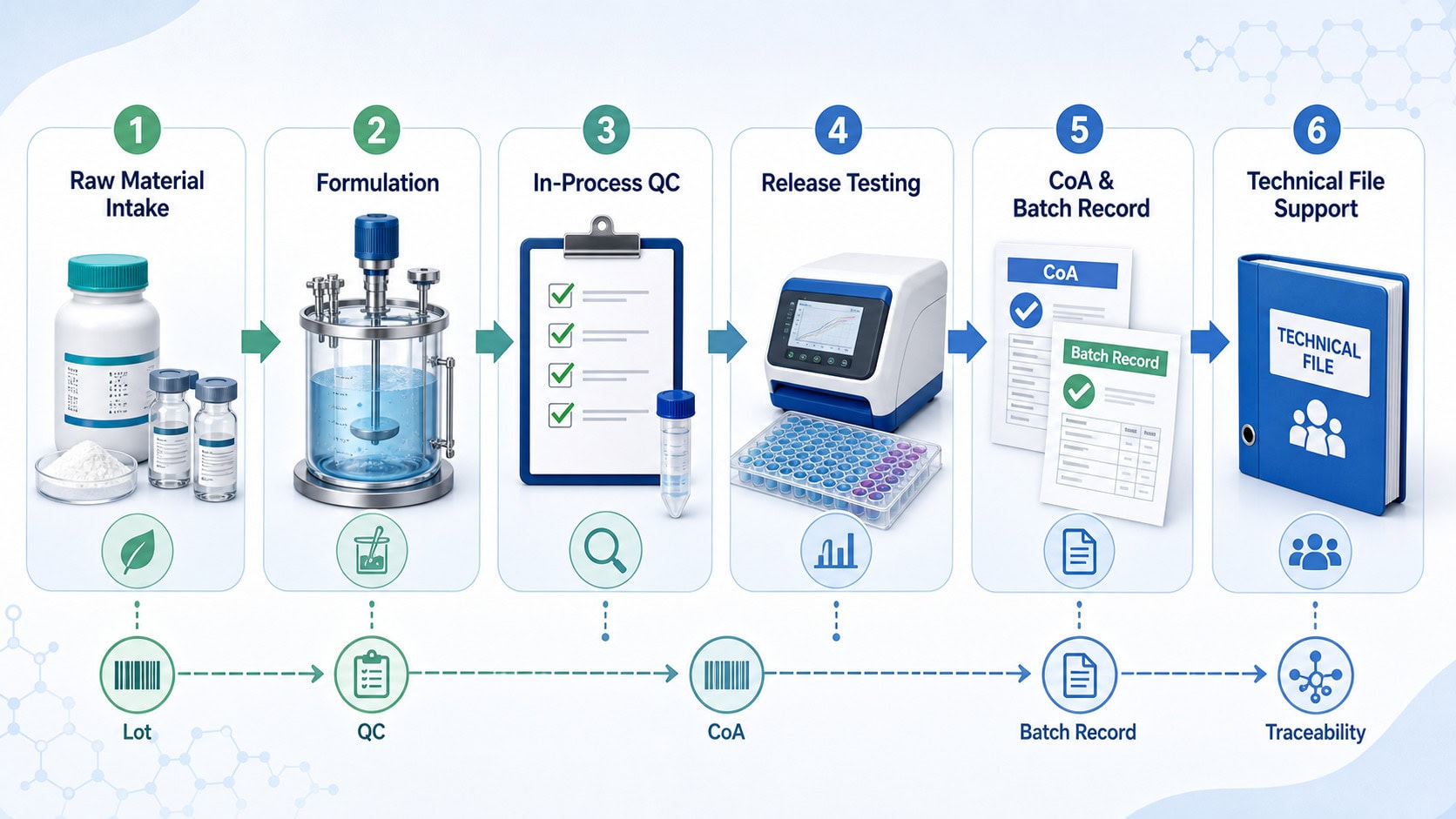
Assay developers should evaluate both the reagent performance data and the documentation system behind the reagent. Depending on the product type, stage of development and regulatory pathway, the useful package may include:
- Raw material controls: supplier qualification, incoming inspection, lot identity and storage records.
- Manufacturing batch records: formulation steps, critical process parameters, operators, equipment and environmental controls where applicable.
- In-process QC: activity, purity, concentration, contamination control, formulation checks and acceptance criteria.
- Release testing: functional assay performance, lot-to-lot consistency, stability or transport-condition evidence when relevant.
- Certificate of Analysis: specifications, test results, lot number, expiry or retest information and quality authorization.
- Traceability support: links among raw materials, intermediate batches, finished lots and customer-facing documents.
- Change-control communication: advance notice and technical rationale for process, specification, supplier or formulation changes.
Documentation Questions to Ask an IVD Reagent OEM Supplier
When screening a supplier for PCR, RT-qPCR, NGS, IVT or biopharma QC reagents, technical teams can use the following questions to reduce downstream uncertainty:
1. Can the supplier map every finished lot to critical raw materials?
Lot traceability is essential when investigating a deviation, complaint or performance shift. Reagent suppliers should be able to connect incoming materials, intermediate production records, finished lots and release documents.
2. Are QC acceptance criteria defined before release?
Release criteria should be specific enough to support consistent lot approval. For enzymes and master mixes, this may include activity, specificity, impurity control, functional amplification performance, residual nuclease or protease risk and formulation stability.
3. Is the CoA useful for technical review?
A CoA should do more than confirm that a lot passed. It should identify the lot, product, specification, tested attributes and release status in a way that supports customer receiving inspection and technical files.
4. Can the supplier support customized documents for OEM programs?
OEM and contract manufacturing projects often need additional documentation, such as formulation records, customer-specific specifications, stability plans, packaging records, change-control agreements and quality questionnaires.
How Documentation Needs Differ by Reagent Type
| Workflow | Typical documentation focus | Customer risk if missing |
|---|---|---|
| PCR / RT-qPCR reagents | Functional amplification, inhibitor tolerance, contamination control, lot consistency | Assay drift, false results, difficult troubleshooting |
| NGS library prep enzymes | Enzyme activity, fragmentation or ligation consistency, impurity control, input compatibility | Library yield variation, bias, failed sequencing runs |
| mRNA / IVT raw materials | GMP-grade enzyme controls, RNase control, residual DNA reduction, capping workflow consistency | Process variability, QC burden, release delays |
| HCD / HCP QC assays | Assay validation support, specificity, sensitivity, standard curve and sample matrix records | Weak residual impurity evidence for biopharma QC |
| Lyophilized or glycerol-free formats | Formulation records, moisture/stability data, packaging and transport-condition evidence | Field instability or poor manufacturing transfer |
How Biori Supports IVD Reagent Quality Documentation
Biori supports overseas customers with biological raw materials and reagent solutions for life science research, molecular diagnostics and biopharma applications. Relevant capabilities include manufacturing and quality systems, OEM and custom manufacturing services, molecular diagnostics reagent selection, lyophilized reagent development and HCD/HCP QC product selection.
For mRNA and IVT-related programs, Biori provides a complete portfolio of GMP-grade IVT raw materials for mRNA manufacturing, including key enzymes and workflow reagents such as T7 RNA Polymerase, Vaccinia Capping Enzyme, 2′-O-Methyltransferase, DNase I, RNase Inhibitor, inorganic pyrophosphatase and related customization support.
For OEM customers, Biori can discuss formulation, scale-up, QC release documentation, lyophilized or glycerol-free options, and technical support packages aligned with customer development stages and quality expectations.
Practical Takeaway for Diagnostic and Biomanufacturing Teams
The current Form 483 guidance discussion is a useful reminder: documentation readiness should be built into supplier selection before a reagent becomes locked into an assay or manufacturing workflow. Teams that evaluate CoA content, batch record structure, traceability, change control and OEM documentation support early can reduce avoidable risk during validation, transfer and commercialization.
If you are developing molecular diagnostic assays, NGS workflows, GMP-grade IVT processes, HCD/HCP QC assays or customized lyophilized reagent formats, contact Biori to discuss enzyme supply, OEM manufacturing and documentation support for your program.
FAQ
What is IVD reagent quality documentation?
IVD reagent quality documentation is the set of records that links raw materials, manufacturing steps, QC testing, release approval, CoA data and traceability for diagnostic reagent lots.
Why does documentation matter for OEM diagnostic reagents?
OEM diagnostic reagents may become part of validated assays or regulated workflows. Clear documentation helps customers qualify suppliers, investigate deviations, manage changes and support technical files.
Should every enzyme supplier provide GMP-grade documentation?
Not every research-use enzyme requires GMP-grade documentation. However, diagnostics, IVT, biopharma QC and late-stage OEM programs often need stronger quality records, traceability and change-control support.
Can lyophilized reagents require additional documentation?
Yes. Lyophilized or glycerol-free reagents may require formulation records, residual moisture or stability evidence, packaging information and transport-condition data to support consistent use.
References
- Fierce Pharma: FDA expectations create potential friction in new Form 483 response guidance
- FDA draft guidance: Responding to FDA Form 483 observations at the conclusion of a drug CGMP inspection
Explore more BIORI technical resources: Browse selection guides, workflow pages, product data and documentation in the BIORI Technical Resources center.
