
Published by Zhuhai Biori Biotechnology Co., Ltd.
Recent biopharma headlines on mRNA manufacturing footprint adjustments, including Fierce Pharma’s report on BioNTech’s manufacturing pullback, are a reminder that RNA programs need more than platform science. A practical mRNA manufacturing raw material strategy helps teams manage process consistency, supplier qualification, documentation, and scale-up risk from early development through routine production.
For organizations developing mRNA vaccines, RNA therapeutics, personalized cancer vaccine workflows, or RNA process tools, the key question is not only whether a raw material works in one experiment. It is whether the enzyme, buffer, capping reagent, nuclease, and QC package can support reproducible manufacturing decisions across multiple batches, sites, and partners.
Why mRNA manufacturing raw material strategy matters now
mRNA manufacturing is highly dependent on specialized biological raw materials. T7 RNA polymerase, capping systems, methyltransferases, DNase, RNase inhibitor, pyrophosphatase, nucleotides, buffers, and purification-related reagents each influence yield, impurity profile, analytical burden, and downstream formulation decisions.
When companies adjust manufacturing networks, consolidate sites, or shift capacity between internal facilities and external partners, raw material planning becomes a central CMC consideration. A well-prepared raw material strategy can help teams:
- reduce lot-to-lot variability in IVT and capping reactions;
- align enzyme quality attributes with the intended stage of development;
- prepare supplier documentation for technical transfer and audits;
- define residual DNA, residual protein, nuclease, endotoxin, and bioburden-related controls;
- support OEM, custom formulation, or lyophilized formats when workflows move between sites.
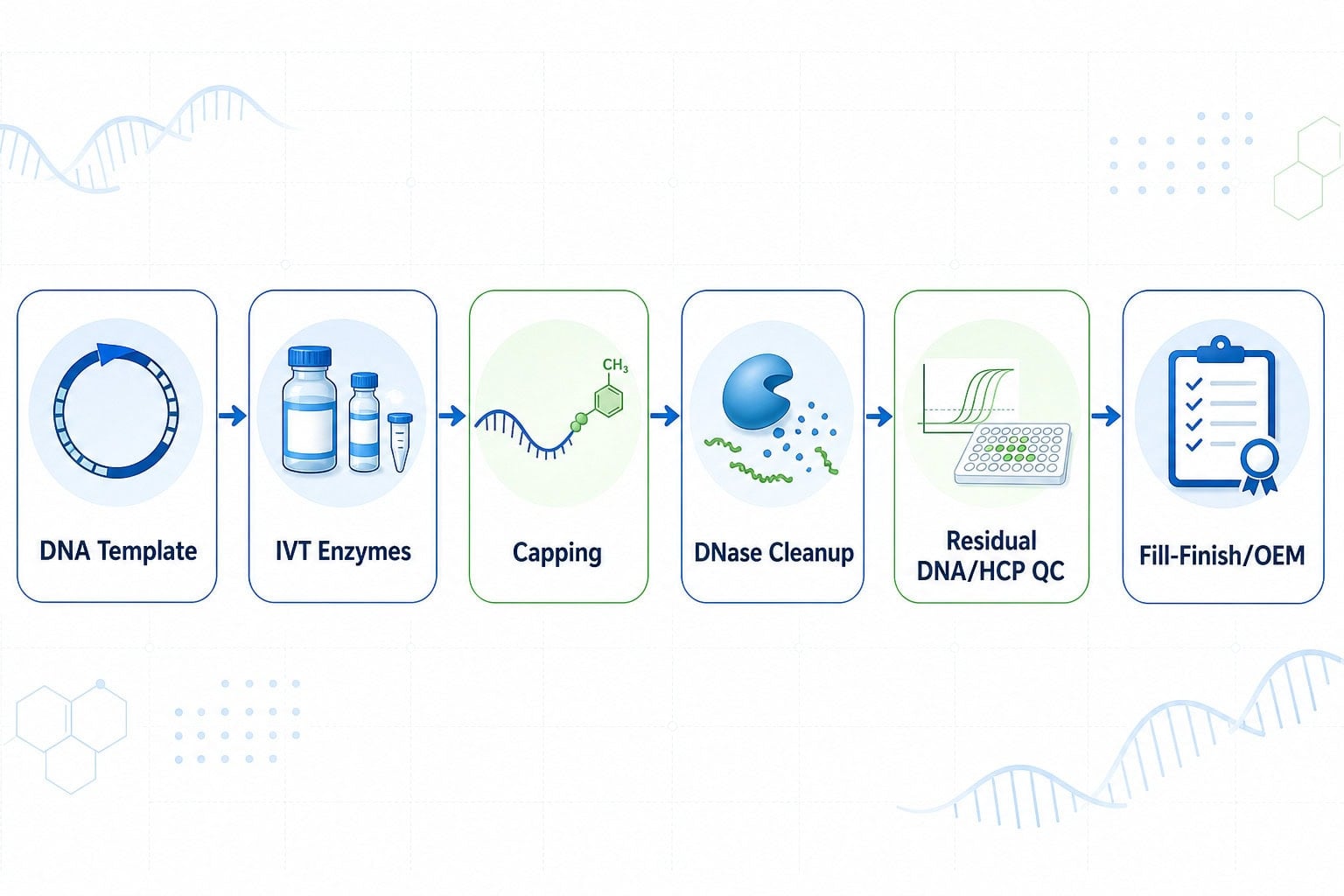
Core raw material categories in an IVT workflow
A resilient mRNA process typically starts with a defined DNA template and then moves through transcription, capping, cleanup, purification, analytics, and final manufacturing documentation. The table below summarizes practical raw material questions for each stage.
| Workflow stage | Typical raw materials | Key questions for sourcing and QC |
|---|---|---|
| DNA template preparation | Restriction enzymes, DNA processing enzymes, buffers | Are digestion efficiency, nuclease contamination, and residual host impurities controlled? |
| In vitro transcription | T7 RNA polymerase, RNase inhibitor, pyrophosphatase, NTPs, reaction buffers | Are activity, purity, dsRNA profile, RNase control, and batch documentation suitable for the program? |
| Capping and methylation | Vaccinia capping enzyme, 2′-O-methyltransferase, SAM, GTP, capping buffers | Is capping efficiency consistent, and can the supplier support scale-up quantities and documentation? |
| DNA cleanup | DNase I or nuclease reagents | Is residual template DNA controlled after digestion and purification? |
| Release and process QC | Residual DNA assays, HCP assays, qPCR/ELISA reagents | Are residual Host Cell DNA and Host Cell Protein risks monitored with appropriate assay strategy? |
Supplier qualification should go beyond activity units
Enzyme activity is important, but activity alone does not define suitability for mRNA manufacturing. Buyers should also evaluate purity, nuclease contamination, host cell impurity controls, endotoxin-related testing, formulation format, storage buffer composition, traceability, and whether the supplier can provide consistent documentation across batches.
For example, a GMP-grade T7 RNA polymerase may be selected not only for high transcription yield, but also for lot release data, residual impurity control, and technical support for modified nucleotide or low-dsRNA workflows. Similarly, capping enzymes and 2′-O-methyltransferases should be assessed for reaction robustness, compatible buffers, scale-up usability, and quality documentation.
QC planning: residual DNA, HCP and process impurities
As mRNA workflows mature, QC teams need to connect upstream raw material decisions with downstream analytical expectations. DNase treatment and purification must be evaluated together with residual DNA testing. Enzyme production systems and recombinant raw materials can also introduce Host Cell Protein considerations, especially for programs moving toward regulated manufacturing environments.
For biopharma QC teams, residual Host Cell DNA (HCD) and Host Cell Protein (HCP) strategies should be discussed early with process development, analytical development, and supplier quality teams. The goal is not to add unnecessary testing, but to create a defensible control strategy that matches the product type, process stage, and risk profile.
How OEM and custom formulation support can reduce transfer risk
Manufacturing footprint changes often create transfer risk: a process optimized at one site must perform similarly at another site, with different equipment, operators, documentation systems, and release timelines. OEM and custom formulation support can help bridge this gap by aligning reagent concentration, buffer system, packaging, fill volume, and quality documentation with the user’s process.
For diagnostic and biopharma customers, lyophilized or glycerol-free formats may also be useful when cold-chain simplification, automation compatibility, or room-temperature handling is important. These formats should be developed with real stability data and application-specific performance testing rather than treated as a simple buffer exchange.
Where Biori fits in the mRNA raw material workflow
Biori provides a complete portfolio of GMP-grade IVT raw materials for mRNA manufacturing, including key enzymes and workflow reagents such as T7 RNA Polymerase GMP-grade, low-dsRNA T7 RNA polymerase, Vaccinia Capping Enzyme GMP-grade, 2′-O-methyltransferase, DNase I, RNase inhibitor, inorganic pyrophosphatase, and restriction enzymes for DNA template preparation.
Biori also supports OEM and custom manufacturing services, lyophilized and glycerol-free reagent development, and HCD/HCP custom assay development for customers building molecular diagnostics, RNA manufacturing, vaccine, antibody, or cell and gene therapy QC workflows.
Practical checklist for mRNA manufacturing raw material strategy
- Map critical raw materials: identify enzymes, buffers, nucleotides, capping reagents, cleanup reagents, and analytical reagents that affect yield or impurity profile.
- Define quality grade by stage: match research, process development, GMP-grade, or custom requirements to the program timeline.
- Review documentation: request CoA, specifications, traceability, storage conditions, and relevant QC test items.
- Plan residual impurity testing: evaluate residual DNA, HCP, nuclease, endotoxin-related and bioburden-related expectations early.
- Evaluate formulation needs: consider glycerol-free, lyophilized, concentrated, or custom buffer formats when workflow transfer or automation is expected.
- Build supplier redundancy carefully: qualify alternates with bridging data rather than relying only on catalog equivalence.
FAQ: mRNA manufacturing raw material strategy
What is the most important enzyme in IVT mRNA manufacturing?
T7 RNA polymerase is usually central to IVT because it drives RNA synthesis from the DNA template. However, capping enzymes, methyltransferases, RNase inhibitor, DNase, and pyrophosphatase can also strongly affect yield, integrity, impurity control, and downstream analytics.
Why are GMP-grade IVT raw materials important?
GMP-grade IVT raw materials support stronger traceability, defined specifications, lot release documentation, and more consistent quality controls. They are especially important when programs move from discovery into process development, clinical manufacturing preparation, or partner technology transfer.
How do residual DNA and HCP testing relate to mRNA manufacturing?
Residual DNA testing helps evaluate removal of DNA template and host-cell DNA impurities. HCP testing can support impurity control for recombinant raw materials or bioprocess inputs. The exact strategy depends on process design, product stage, and regulatory expectations.
Can mRNA reagents be customized for OEM manufacturing?
Yes. OEM programs may require custom concentration, buffer composition, glycerol-free formulation, lyophilization, packaging, documentation, or QC release criteria. Early technical discussion helps ensure the reagent format matches the customer’s process and instruments.
References
- Fierce Pharma: BioNTech manufacturing footprint report
- FDA CBER: development and approval process information for biologics
Talk to Biori if your team is evaluating GMP-grade IVT enzymes, mRNA capping and cleanup reagents, HCD/HCP QC assays, OEM manufacturing, or lyophilized/glycerol-free formats for international RNA, IVD, vaccine, antibody, or CGT programs.
